
A 21-year-old woman with a history of sickle cell disease (SCD) presented to the emergency department with complaints of bilateral vision loss. The patient reported a complete blackout in both eyes that lasted approximately 1 to 2 hours while she was drinking alcohol 3 days ago. Since then, her vision had gradually improved but had not returned to baseline.

Figure 1. Ultra-widefield fundus photography of the right (A) and left (B) eye demonstrated retinal whitening secondary to CRAO.
EXAMINATION
On examination, her VA was counting fingers OD and 20/25 OS. IOP was within normal limits. No relative afferent pupillary defect was noted. Anterior segment examination was unremarkable. Dilated fundus examination of each eye revealed a cherry red spot and retinal edema consistent with concurrent central retinal artery occlusion (CRAO; Figure 1). OCT imaging showed inner- and middle-layer hyperreflectivity and thickening (Figure 2).
The patient’s arterial occlusions were thought to be secondary to her SCD, and she was admitted for emergent exchange transfusion. A stroke and hypercoagulability workup showed low protein C, protein S, and antithrombin III levels and elevated lipoprotein A and factor VIII. The hemoglobinopathy panel indicated elevated hemoglobin S (HbS) levels (90.7%). The day following the exchange transfusion, the patient’s VA improved to 20/400 OD, and the left eye remained stable. The repeat hemoglobinopathy panel showed decreased HbS levels (19.5%). Given the patient’s hypercoagulability workup and that her mother had a history of deep vein thrombosis and pulmonary embolism, the patient was started on anticoagulation.
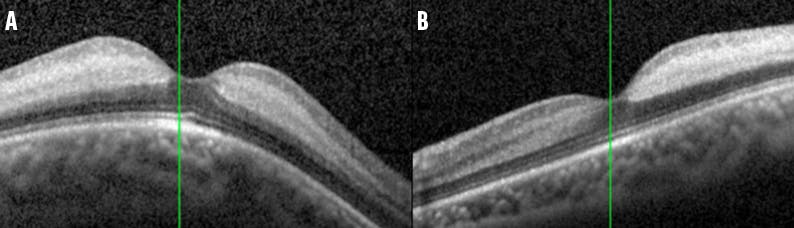
Figure 2. OCT of the right (A) and left (B) eye showed increased reflectivity and thickening of the inner retinal layers consistent with CRAO.
DISCUSSION
Sickle cell retinopathy (SCR) is the most common manifestation of SCD and can be classified as nonproliferative (NPSCR) or proliferative (PSCR). The retinal changes in NPSCR are secondary to vascular occlusion and local ischemia.1 Clinical findings include peripheral retinal vascular anastomoses, salmon patches, iridescent spots, and black sunbursts. Salmon patches are well-demarcated, superficial intraretinal or preretinal hemorrhages, whereas iridescent spots represent hemosiderin-filled macrophages in areas of old, resorbed hemorrhages and within the inner retina just beneath the internal limiting membrane. Sunburst lesions appear after resolution of the hemorrhages and are localized areas of retinal pigment epithelium hyperplasia and pigment migration. Central retinal changes can consist of arteriovenous tortuosity, foveal avascular zone enlargement, and arterial occlusions.1,2 A foveal depression sign has also been described, characterized as a darkened foveal reflex due to thinning of the retinal layers in this region.3
PSCR is characterized by the development of peripheral retinal neovascularization, the hallmark sign being a sea fan configuration.1,2 Sea fans have a high propensity to regress by autoinfarction, although they may lead to vision loss through vitreous hemorrhage or tractional retinal detachment.2 Genotype is the risk factor most strongly associated with the development of PSCR.
Vision-threatening PSCR occurs earlier and is more likely to affect those with hemoglobin C SCD (HbSC) versus homozygous SCD (HbSS).4 Several theories have been proposed for this, including higher hematocrit levels in HbSC, which may lead to increased red blood cell sludging and small vessel occlusion in the retinal vasculature.5 Another theory is that vascular occlusion in HbSC may be less severe, resulting in low-grade chronic ischemia and release of proangiogenic growth factors, which creates a more favorable environment for neovascularization. In contrast, the vascular occlusion in HbSS is more complete, resulting in more profound infarction and retinal necrosis, which is less likely to generate an angiogenic response.5
Bilateral concurrent CRAO secondary to SCD is extremely rare. To the best of our knowledge, only three cases have been previously reported.6-8 The exact pathogenesis of vascular occlusion in SCD is poorly understood, but several mechanisms have been proposed, including abnormal endothelial adhesion by sickled erythrocytes, activation of the coagulation cascade, and intimal injury. Renganathan et al reported a case of a 24-year-old woman with a history of SCD who presented with a VA of hand motion OU in the setting of CRAO during a sickle crisis. Immediate exchange transfusion was initiated and continued until her symptoms improved over 2 days.6 Murthy et al similarly reported a case of a 37-year-old woman who presented with a VA of no light perception OU and had recently been started on a phosphodiesterase type 5 inhibitor for pulmonary hypertension. Although immediate exchange transfusion was initiated, the patient did not recover any vision.7
In our case, we believe that alcohol may have precipitated a sickle crisis, leading to bilateral occlusion of the central retinal artery.
The mainstay for treatment in SCD remains hydroxyurea or blood transfusion. Transfusions help to decrease the proportion of red sickle cells in circulation while simultaneously improving anemia and peripheral tissue oxygen delivery. For acute SCD complications, the goal of transfusion therapy is to reduce the posttransfusion HbS level to less than 30%.
WHILE RARE, BE AWARE
Concurrent retinal arterial occlusion in SCD is exceedingly rare. Exchange transfusion should be initiated emergently in cases that do present to attempt to reverse some of the vision loss in these patients.
1. Abdalla Elsayed MEA, Mura M, Al Dhibi H, et al. Sickle cell retinopathy. A focused review. Graefes Arch Clin Exp Ophthalmol. 2019;257(7):1353-1364.
2. Scott AW. Ophthalmic manifestations of sickle cell disease. South Med J. 2016;109(9):542-548.
3. Lim JI. Sickle cell retinopathy. In: Albert DM, Miller JW, Azar DT, Young LH, eds. Albert and Jakobiec’s Principles and Practice of Ophthalmology. Springer International Publishing; 2022:3103-3123.
4. Fox PD, Dunn DT, Morris JS, Serjeant GR. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol. 1990;74(3):172-176.
5. Fadugbagbe AO, Gurgel RQ, Mendonça CQ, Cipolotti R, dos Santos AM, Cuevas LE. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010;30(1):19-26.
6. Renganathan G, Natarajan P, Ruck L, Prieto R, Prakash BV, Thangarasu S. Concurrent bilateral central retinal artery occlusion secondary to sickle cell crisis. J Invest Med High Impact Case Rep. 2021;9:23247096211028392.
7. Murthy RK, Perez L, Priluck JC, Grover S, Chalam KV. Acute, bilateral, concurrent central retinal artery occlusion in sickle cell disease after use of tadalafil (Cialis). JAMA Ophthalmol. 2013;131(11):1471-1473.
8. Goodwin PL, Vaphiades MS, Johnson AP, Stroud CE. Bilateral central retinal artery occlusion associated with Moyamoya syndrome in a sickle cell disease patient. Neuroophthalmol. 2008;32(1):21-26.


_1784132761.jpg?auto=compress,format&w=75)











