

Bardet-Biedl syndrome (BBS) is a rare, autosomal recessive, inherited systemic disease categorized as a nonmotile ciliopathy. Mutations in 26 genes have been identified to cause BBS, with BBS1 being the most frequent (23%), followed by BBS10 (15%) and BBS2 (10%).1,2 Rods and cones contain the primary cilia-like structures affected by such mutations; thus, this disease process hinders protein transport and leads to their intracellular accumulation, photoreceptor death, and vision loss.2 Here, we describe the findings of three siblings diagnosed with BBS.
CASE REPORT
A 20-year-old woman presented to our clinic with nyctalopia and a gradual decrease in vision. At presentation, her BCVA was 20/50 OD and 20/80 OS. She had high myopia and astigmatism. Past medical history included obesity since childhood, operated polydactyly, polycystic ovary syndrome, and a learning disability. Her twin brother and a younger brother each had polydactyly, obesity, and learning disabilities as well. She also had a paternal aunt with a history of polydactyly and death from ovarian cancer at age 27, and her maternal grandfather and aunt had severe diabetes mellitus.
Slit-lamp examination was normal. Color fundus imaging and fundus autofluorescence (FAF) revealed central macular and peripheral pigmentary changes in each eye (Figure 1A and B). Spectral-domain OCT (SD-OCT) demonstrated outer retinal disruption (Figure 1C).
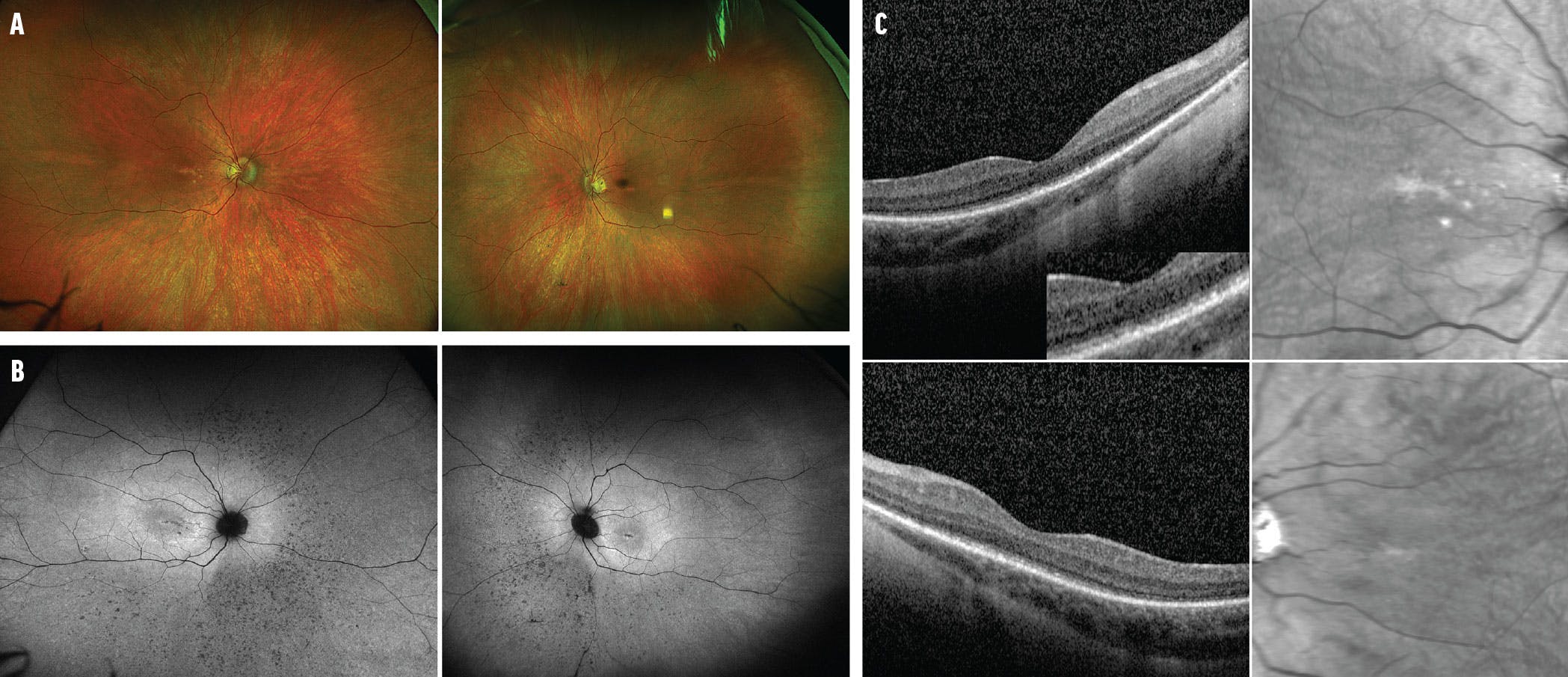
Figure 1. Fundus photography of a 20-year-old woman shows pigmentary changes in the macula and periphery of the right and left eyes (A). FAF shows diffuse retinal pigmentary changes in the right and left eyes (B). SD-OCT demonstrates ellipsoid zone disruption in the right and left eyes (C).
The patient’s twin brother had a history of operated polydactyly, obesity, more severe cognitive impairment, high myopia, and astigmatism. His BCVA was 20/30 OU, tested with Allen pictures blocked. Color fundus imaging and FAF revealed pigmentary changes in the macula, along with more extensive peripheral pigmentary changes compared with his sister (Figure 2A and B). SD-OCT demonstrated marked outer retinal disruption (Figure 2C).
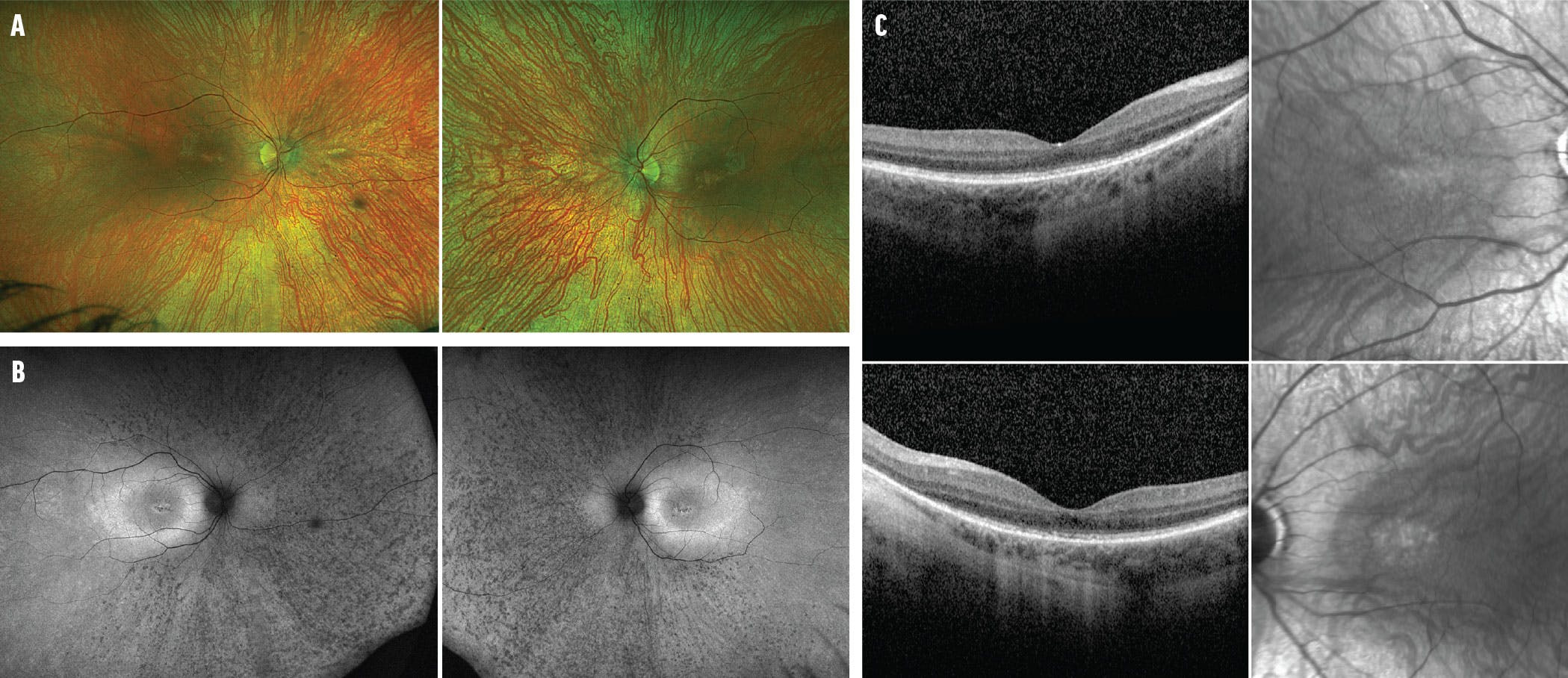
Figure 2. Fundus photography of the right and left eyes of the twin brother shows macular and peripheral pigmentary changes (A). FAF of the right and left eyes shows diffuse retinal pigmentary changes (B). SD-OCT of right and left eyes demonstrates ellipsoid zone disruption (C).
The twins’ 17-year-old brother had a history of operated polydactyly, obesity, and mild cognitive impairment, along with moderate myopia and astigmatism. His BCVA was 20/40 OD and 20/30 OS. Color fundus imaging and FAF revealed a milder pigmentary retinopathy compared with his siblings and a hyperautofluorescent lesion in the macula of each eye (Figure 3A and B). SD-OCT imaging demonstrated subretinal deposits in the fovea corresponding with the hyperautofluorescent lesions, which are likely a precursor to future photoreceptor disruption and degeneration (Figure 3C).

Figure 3. Fundus photography of the right and left eyes of the 17-year-old brother shows more subtle pigmentary changes compared with his older siblings (A). FAF of the right and left eyes shows a subtle pigmentary retinopathy and a hyperautofluorescent macular lesion (B). SD-OCT demonstrates subretinal deposits in the fovea of each eye (C).
Genetic Testing
The three siblings underwent genetic testing, which revealed a homozygous pathogenic BBS1 gene with a c.1169T>G (p.Met390Arg) variant in each sibling. This sequence change replaces methionine with arginine at codon 390 of the BBS1 gene. The genotype matches the phenotype.
Genetic testing established a diagnosis of BBS for all three siblings. Given their obesity, risk of diabetes, and cognitive impairment, they were referred to their pediatrician for a renal, cardiac, neurological, and genitourinary evaluation.
DISCUSSION
The prevalence of BBS is estimated to be between 1 in 140,000 and 1 in 160,000 newborns in North America and Europe, with 44 new cases reported annually in the United States.3 Its most exhibited features are retinal rod-cone dystrophy with variable presentations (94% of cases), obesity and related complications, cognitive delays, and genitourinary and renal dysfunction.1Retinal dystrophy can range from subtle macular pigmentary changes to bull’s eye maculopathy, along with peripheral pigmentary changes with atrophy and bone spicules.1,3
Patients usually present in the first decade of life with symptoms of nyctalopia, gradual loss of peripheral vision, color discrimination deficits, and decreased visual acuity. Affected individuals can experience significant vision loss in the third decade of life. Strabismus, astigmatism, and cataracts may also be found.1
Other systemic features include brachydactyly; syndactyly; deficits in hearing and smell; facial, cranial, and dental dysmorphia; and gastrointestinal, skin, and renal diseases. Neurologic findings, such as seizures, ataxia, and developmental delays (speech and behavior), are also present. BBS shows a range of expressivity that can vary between families and individuals within the same family.1
Diagnosing BBS
The differential diagnosis of BBS is comprised of various syndromic pigmentary retinopathies, including ciliopathies (ie, Joubert syndrome, characterized by brain abnormalities, and Senior Loken syndrome, characterized by progressive kidney disease), Refsum syndrome, familial isolated vitamin E deficiency, and Alström syndrome, among others.1,4,5 It is the second most common autosomal recessive syndromic ciliopathy, after Usher syndrome, which is also known to cause sensorineural hearing loss.1,5
High suspicion of BBS is inferred from personal and family history and clinical findings; however, genetic testing and counseling are required to confirm the diagnosis.1,4,6 Given its widespread systemic involvement, obtaining an accurate diagnosis is paramount in the management of childhood obesity, diabetes mellitus, kidney dysfunction, and heart defects that can become life-threatening.1
Treatment Approaches
Management of BBS is primarily aimed at reducing symptoms and focuses on addressing the individual’s specific needs. Multidisciplinary care involving various specialists may be necessary, including low vision services.1 In 2020, the FDA approved setmelanotide injections (Imcivree, Rhythm Pharmaceuticals) for progressive weight gain management in patients with BBS older than 6 years of age. The results of a 66-week trial that enrolled 44 individuals with BBS and obesity showed that treatment with setmelanotide led to an average decrease in body mass index of 7.9%.7,8
Many genetic syndromes have systemic complications that can be progressive and debilitating if left untreated. Identifying BBS early allows for prompt initiation of appropriate medical interventions and therapies.
STRIVE FOR BETTER OUTCOMES
Early syndrome recognition and genetic testing are important steps in the diagnosis and management of genetic disorders such as BBS and can lead to better outcomes and improved quality of life. Testing also enables timely referrals to specialists, informs family planning, and allows earlier access to treatments and participation in clinical trials.
1. Forsyth R, Gunay-Aygun M. Bardet-Biedl syndrome overview. In: GeneReviews. Seattle: University of Washington, Seattle; 1993.
2. Weihbrecht K. Bardet-Biedl syndrome. In: Genetics and Genomics of Eye Disease. Academic Press; 2020:117-136.
3. Bardet-Biedl syndrome. Medline Plus. Accessed September 27, 2023. medlineplus.gov/genetics/condition/bardet-biedl-syndrome
4. Tsui I, Song BJ, Lin CS, Tsang SH. A practical approach to retinal dystrophies. Adv Exp Med Biol. 2018;1085:245-259.
5. Cantani A, Bellioni P, Bamonte G, Salvinelli F, Bamonte MT. Seven hereditary syndromes with pigmentary retinopathy: a review and differential diagnosis. Clinical Pediatrics. 1985;24(10):578-583.
6. Hartong DT, Berson EL, Dryia TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
7. FDA approves treatment for weight management in patients with Bardet-Biedl Syndrome aged 6 or older. US FDA. June 16, 2022. Accessed September 29, 2023. www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-weight-management-patients-bardet-biedl-syndrome-aged-6-or-older
8. Setmelanotide (RM-493), melanocortin-4 receptor (MC4R) agonist, in Bardet-Biedl Syndrome (BBS) and Alström Syndrome (AS) patients with moderate to severe obesity. Clinicaltrials.gov. Updated June 20, 2021. Accessed September 29, 2023. classic.clinicaltrials.gov/ct2/show/NCT03746522







_1784132761.jpg?auto=compress,format&w=75)











